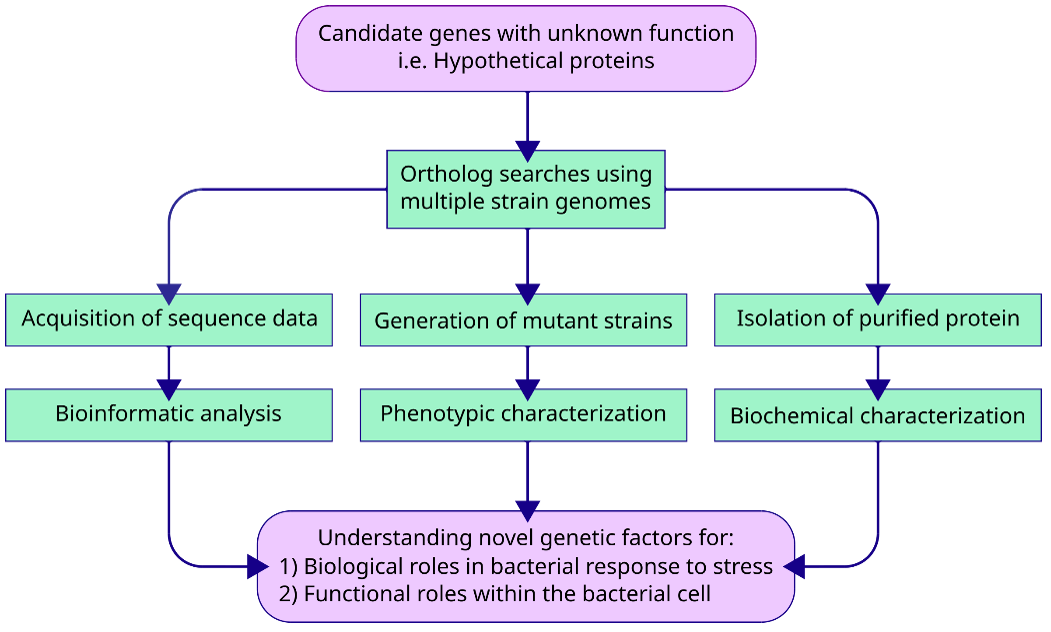
Since the completion of the first bacterial genome in 1995, we have seen massive technological advances and cost reduction in genome sequencing over the last 25 years. This has resulted in a large number of available bacterial genomes (263,273 bacterial genomes available on NCBI as of 2020/09/19). However, our ability to sequence a bacterial genome is much faster than we can functionally characterize the genome. For instance, even among the best-studied model organism, Escherichia coli K-12 MG1655, ~35% of its genome has poor functional annotations. These poorly annotated genes, typically named as “hypothetical proteins”, are propagated across genome databases as standard bioinformatic pipelines cannot predict their functions. Given that many of these hypothetical proteins play a critical role in host-pathogen interactions, we have developed a prioritization strategy to identify candidate genes for functional characterization. Specifically, we propose to: (1) perform bioinformatic data analyses to identify conserved hypothetical proteins that are associated with resistance or virulence phenotypes; (2) validate these phenotypes experimentally in the lab using deletion mutants; (3) perform protein expression, purification and biochemical characterization of interesting candidates. Current pathogens of interest include Pseudomonas aeruginosa, Burkholderia cepacia complex, and Klebsiella pneumoniae.